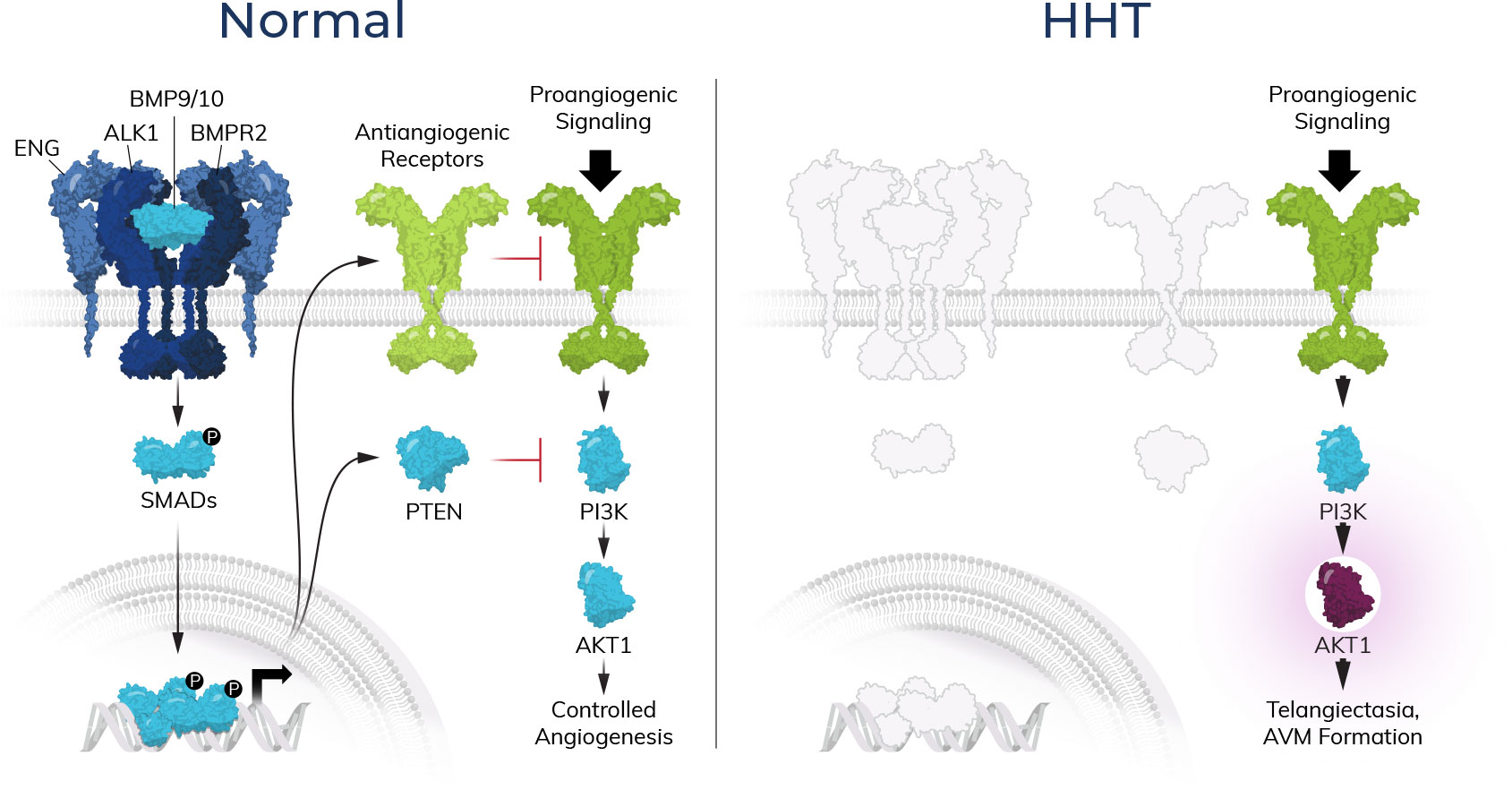
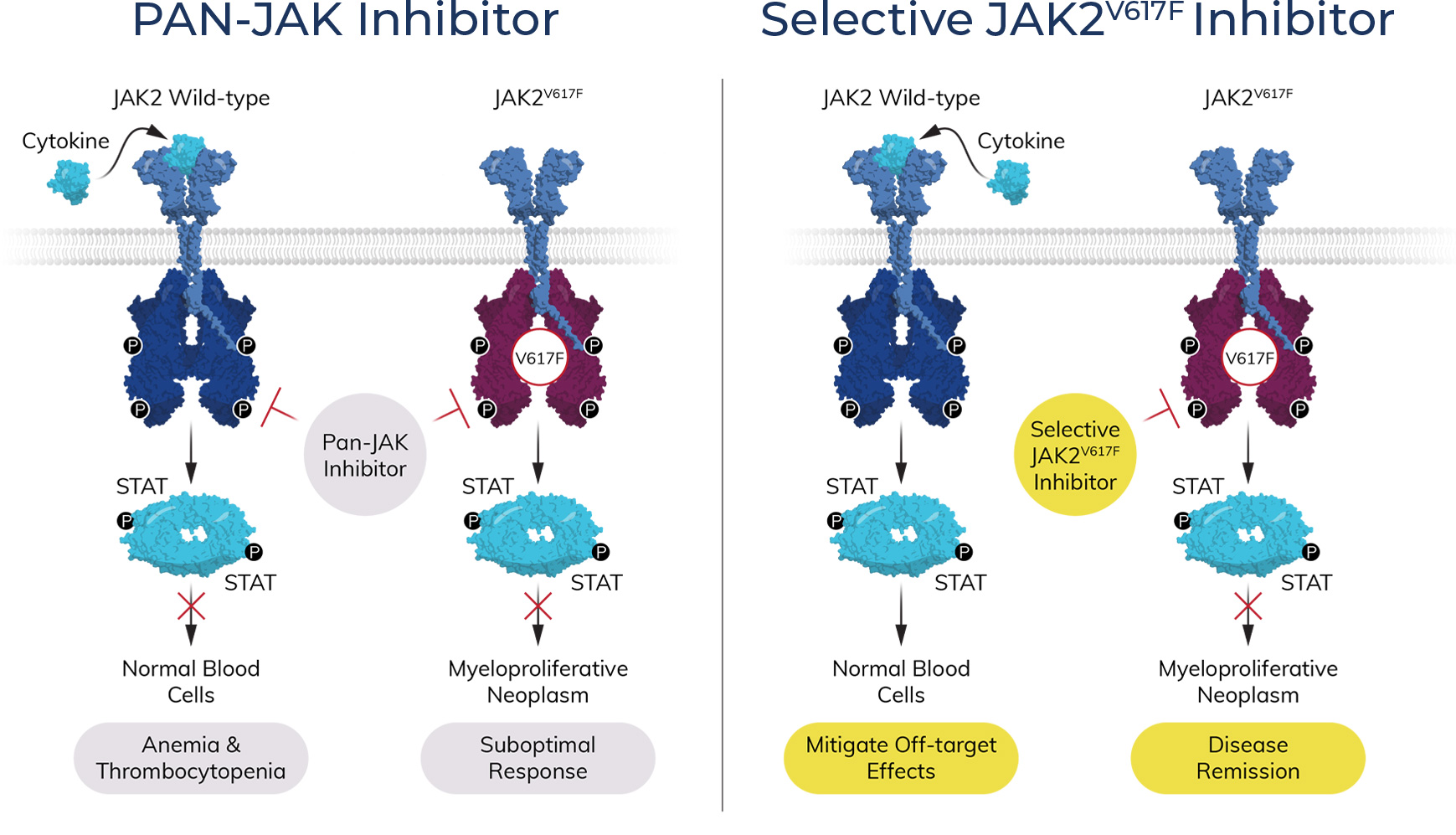
At Atavistik Bio, we are developing a pipeline of transformative allosteric therapeutics to address serious unmet patient needs. Our AMPS™ platform has enabled us to efficiently deliver best-in-class small molecule allosteric inhibitors for high value targets, with an internal focus on rare hematology.